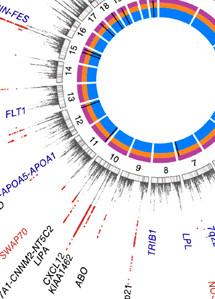
Genome wide association studies (GWAS) have greatly advanced our understanding of the genetics of complex diseases such as coronary artery disease, diabetes, and hypertension. In some instances, the identification of SNPs in genomic regions, which point to potential disease-related genes, led to the identification of new therapeutic targets.
However, an extremely large proportion of published GWAS has focused on the analysis of the 22 autosomal chromosomes only. As a consequence, although the X chromosome constitutes 5% of the nuclear genome and underlies almost 10% of Mendelian disorders, it harbors only 15 of the 2,800 (0.5%) significant associations reported by GWAS of nearly 300 traits.
GWAS findings mainly concern autosomal chromosomes and rarely the X chromosome. Indeed, the X chromosome is commonly excluded from GWAS analyses despite being assayed for a limited number of SNPs by all current GWAS microarray platforms.
There are various reasons for not including the X chromosome in GWAS: i) poor coverage of the X chromosome, ii) increased workload owing to gender-specific quality controls, iii) power issues owing to a smaller sample size, and most importantly iv) the requirement for specific tools to analyze X chromosomal data.
Such specific tools are needed because of the characteristics of the X chromosome that make it distinct from autosomal chromosomes. First, we need to consider the unique genetic make-up of the X chromosome; females have two copies, while males have one copy, excluding the pseudo-autosomal regions. Thus, when males are included in analyses, variants on X chromosomal loci require special treatment. For example, the signal intensities obtained from standard array genotyping platforms are lower for males, who carry one allele, than for females, who carry two alleles. This needs to be adequately addressed in the genotype-calling step and has consequences for genotype imputation and association analyses.
Second, we need to consider the process of X chromosomal inactivation. Early in embryonic development, large parts of one of the two female X chromosomes are silenced, which is hypothesized to occur via dosage compensation. This results in the one copy of the X chromosome in males and the two copies of the X chromosome in females having equal effects. This inactivation is incomplete, and it is estimated that about three-quarters of X chromosomal genes are silenced in one female X chromosomes in some individuals. This is important when deciding how to test for associations with X chromosomal variants as described in a recent study by König et al. 2014.
To overcome the hurdles of X chromosomal analyses, Inke König and colleagues have established pipelines for analyzing X chromosomal data within a standard GWAS. By selecting specific algorithms and parameter settings, the analysis of X chromosomal SNPs is manageable and gives new clues as to the genetics of complex diseases.
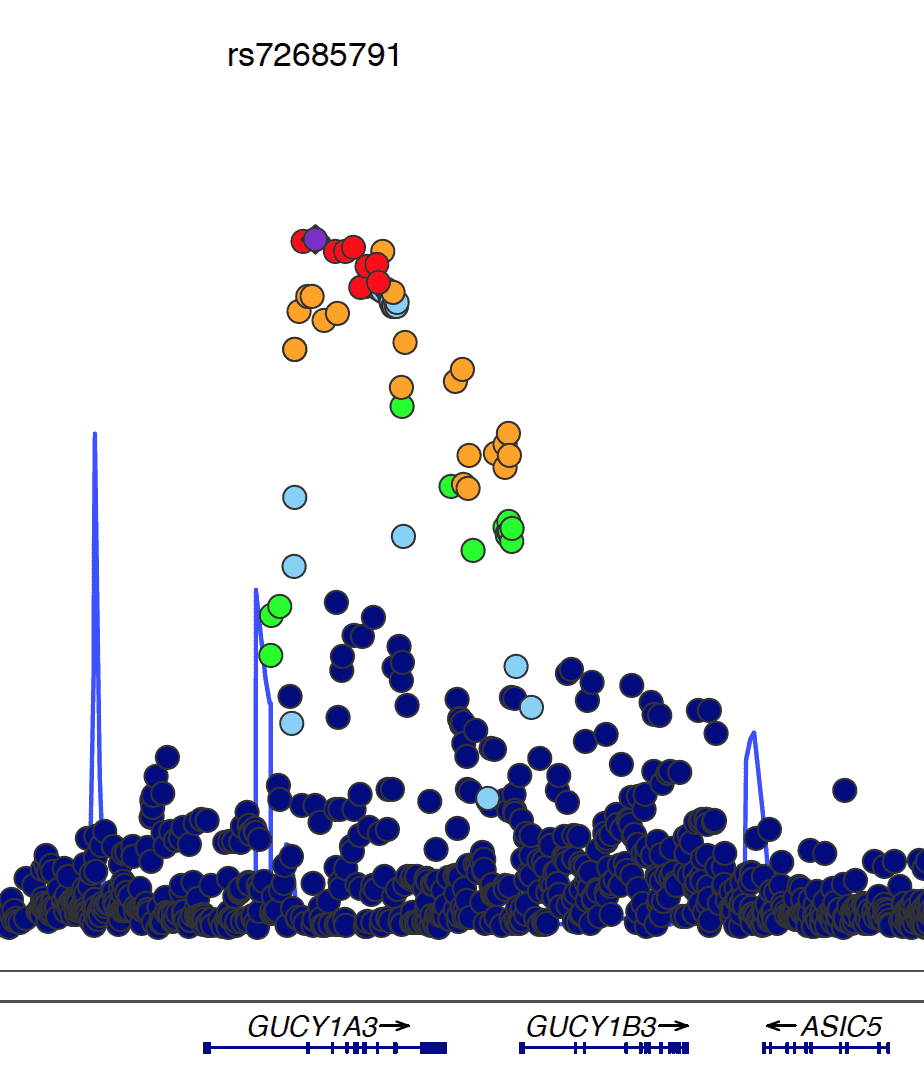
Here, we now present the very first comprehensive meta-analysis of SNPs located on X chromosome. We expected that inclusion of X chromosomal data might partly explain the so-called missing heritability of complex diseases, especially those with sex-specific features. However, although we analyzed the largest-to-date sample, currently available methods were not able to detect any associations of X-chromosomal variants with CAD.
link to open access article

 ting talks from our bachelors, masters, PhD/MD and postdocs. In between there was ample time for leisure, like bosseln, and in the evening we cooked under guidance a delicious Dinner.
ting talks from our bachelors, masters, PhD/MD and postdocs. In between there was ample time for leisure, like bosseln, and in the evening we cooked under guidance a delicious Dinner.